
Genozip Enterprise
Objective
Genozip Enterprise is designed for medium-to-large deployments ranging from 50 TB to petabytes, and contains our best compression methods, plus, of course, everything in Genozip Standard.
The incremental functionality available in Genozip Enterprise which is not available in Genozip Standard, consists of three compression-enhancing methods: Deep, Pair and Optimize.
These options are also available in Genozip Premium.
Deep: co-compression of FASTQ and BAM

Genozip Deep™ is a patent-pending method for co-compressing a BAM file (or SAM or CRAM) along with all the FASTQs that contributed reads to the BAM. The result of this compression is a single file with the extension .deep.genozip. Uncompressing the deep file with genounzip outputs all the original files (losslessly, obviously!). This method results in very substantial savings vs compressing the FASTQs and the BAM separately, as can be appreciated from the benchmark above.
The exact compression ratios achieved are very much dependent on the specifics of the data being compressed, but it is not unusual to achieve file size reductions of 85%-90% with this method.

Pair: co-compression of paired-end FASTQ files
The left bar shows sizes of each .fastq.genozip file when compressed separately, relative
to the combined size of the .fastq.gz files, and the right bar shows the relative size of the
.fastq.genozip file when co-compressed together using --pair.
With the --pair option, Genozip exploits redundancies between corresponding reads in a pair of FASTQ files to improve compression when they are compressed together. Typically, this results in shrinking the compressed file by an additional 10-15%.
By default, decompression recovers the original two FASTQs, however, it is also possible to output them together in interleaved format.
Optimize: even better compression, with some caveats.
While Genozip is primarily a lossless compressor, the --optimize option is where we venture into "lossy compression" as well. The idea of lossy compression is this: It is common for data to contain information in a higher resolution than we actually need for downstream analysis. Examples include resolution of base quality scores and number of decimal digits in fractions. If we could reduce the resolution, we could gain signficantly better compression. The compression-enhacing modifications which genozip performs when --optimize is used, are designed to have negligible impact on downstream analysis in many common cases, however you should validate this for your own data.
The additional savings with the Optimize method highly depend on the details of the specific file being compressed. If the file is already highly optimized, then --optimize might have limited effect. In other files, it might halve the size of the file or better, compared to genozip without --optimize.
Those who resent the Z in the word "optimize" will be glad to know that --optimise works as well 😊.
Details of the specific modifications can be found here: FASTQ SAM/BAM/CRAM VCF
Optimize for FASTQ
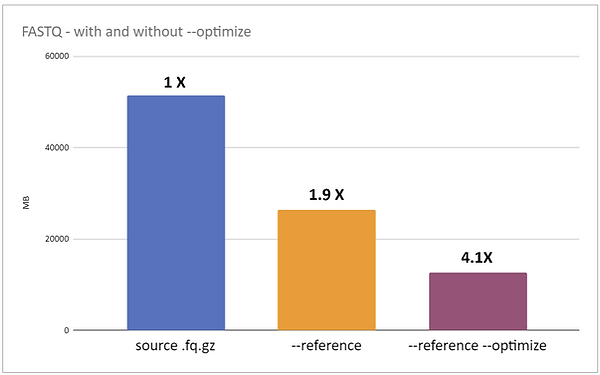
Genozip compression without and with --optimize, showing file sizes in MBs. The file tested is a FASTQ file produced by an MGI Tech sequencer, obtained from here.
Optimize for BAM

Genozip compression without and with --optimize, showing file sizes in MBs. The file tested is a BAM file which consists of MGI Tech reads aligned with Illumina DRAGEN.
Optimize for VCF

Genozip compression without and with --optimize, showing file sizes in MBs. The file tested is a GVCF file produced by Illumina DRAGEN obtained from here.
Questions? support@genozip.com